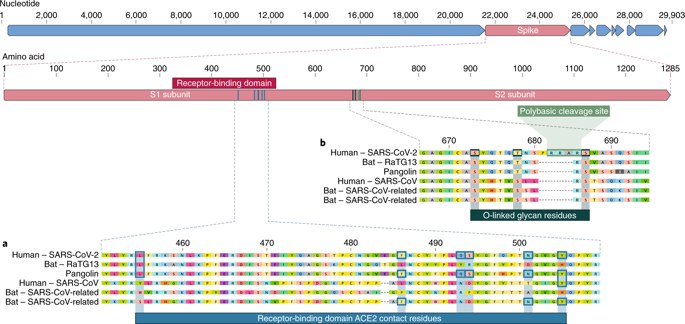
Notable features of the SARS-CoV-2 genome
Our comparison of alpha- and betacoronaviruses identifies two notable genomic features of SARS-CoV-2: (i) on the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 appears to be optimized for binding to the human receptor ACE2; and (ii) the spike protein of SARS-CoV-2 has a functional polybasic (furin) cleavage site at the S1–S2 boundary through the insertion of 12 nucleotides8, which additionally led to the predicted acquisition of three O-linked glycans around the site.
1. Mutations in the receptor-binding domain of SARS-CoV-2
The receptor-binding domain (RBD) in the spike protein is the most variable part of the coronavirus genome1,2. Six RBD amino acids have been shown to be critical for binding to ACE2 receptors and for determining the host range of SARS-CoV-like viruses7. With coordinates based on SARS-CoV, they are Y442, L472, N479, D480, T487 and Y4911, which correspond to L455, F486, Q493, S494, N501 and Y505 in SARS-CoV-27. Five of these six residues differ between SARS-CoV-2 and SARS-CoV (Fig. 1a). On the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 seems to have an RBD that binds with high affinity to ACE2 from humans, ferrets, cats and other species with high receptor homology7.
Fig. 1: Features of the spike protein in human SARS-CoV-2 and related coronaviruses.

a, Mutations in contact residues of the SARS-CoV-2 spike protein. The spike protein of SARS-CoV-2 (red bar at top) was aligned against the most closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues in the spike protein that make contact to the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in lineage B betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146 and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378(NCBI BioProject PRJNA573298)29,30.
Full size image
While the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal7 and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding7,11. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation.
2. Polybasic furin cleavage site and O-linked glycans
The second notable feature of SARS-CoV-2 is a polybasic cleavage site (RRAR) at the junction of S1 and S2, the two subunits of the spike8 (Fig. 1b). This allows effective cleavage by furin and other proteases and has a role in determining viral infectivity and host range12. In addition, a leading proline is also inserted at this site in SARS-CoV-2; thus, the inserted sequence is PRRA (Fig. 1b). The turn created by the proline is predicted to result in the addition of O-linked glycans to S673, T678 and S686, which flank the cleavage site and are unique to SARS-CoV-2 (Fig. 1b). Polybasic cleavage sites have not been observed in related ‘lineage B’ betacoronaviruses, although other human betacoronaviruses, including HKU1 (lineage A), have those sites and predicted O-linked glycans13. Given the level of genetic variation in the spike, it is likely that SARS-CoV-2-like viruses with partial or full polybasic cleavage sites will be discovered in other species.
The functional consequence of the polybasic cleavage site in SARS-CoV-2 is unknown, and it will be important to determine its impact on transmissibility and pathogenesis in animal models. Experiments with SARS-CoV have shown that